An introduction to scRNA-seq pt. 1
Intro

There are tons of different sequencing technologies.
It seems like these days anything can be sequenced, and your expected to not only know each sequencing technique and methodology, but also how to combine data from different methods and know how to analyze it in detail.
But, as the old adage goes, the journey of a thousand miles begins with a single step. So today we are going to begin the first of a 3-part series exploring-
Single-cell RNA-Sequencing 🧬
This article will be followed up by 2 more-
- Processing scRNA-seq data.
- Analyzing scRNA-seq data.
As usual, this article will focus on what we data scientists call the intuition and try and answer 3 main questions at a high level-
- What is scRNA-seq?
- How are the samples prepared?
- What are the differences between scRNA-seq and bulk RNA-seq?
Let’s get started-
Overview of scRNA-seq
Much of what scRNA-seq can be conveniently intuited by the name single-cell RNA sequencing. We can sequence the RNA transcripts in a single cell.
This allows us to get gene expression levels from a single cell.
Contrast this with more widely understood bulk RNA-seq, which allows a researcher to get the gene expression levels from a tissue sample containing multiple cell types.
For example, say I want to investigate the effect of a drug on gene expression in skeletal muscle in obese patients vs. patients with normal weight.
I would probably take muscle biopsies from some region (e.g. the vastus lateralis/the outer thigh), lyse the tissue, and sequence the RNA-transcripts using bulk RNA-sequencing.
While this would give you gene expression, we are unable to see which genes in specific cell types in particular changed their expression levels.
This is because our muscle biopsy is composed of different cells, not just skeletal muscle. There could be stem cells, muscle cells, adipocytes, blood cells, and skin cells all in a single muscle biopsy.
And because we lysed and sequenced it all together without differentiation, there’s no way to see which cell types responded to the drug.
In short, bulk RNA-seq allows a researcher to measure gene expression at the level of the tissue while with scRNA-seq allows are researcher to get gene expression at the level of the cell. ✅
Let’s explore how this is accomplished.
Microfluidic-droplet based methods (10x Genomics)
There are 3 common ways to conduct scRNA-seq
-
Microtitre-plate methods
-
Microfluidic-array methods
-
Microfluidic-droplet methods
Microfluidic-droplet is the highest throughput and the most common, so we will focus on understanding this method.
The high level steps are as follows-
- Obtain sample from populations of interest.
i.e. taking our skeletal muscle biopsies from our obese vs. normal weight groups.
-
Mechanically or enzymatically disaggregate your sample and suspend in solution.
-
Capture individual cells in oil droplets.
During this step, the individual cells are captured in tiny oil droplets that contain a bead. The bead contains enzymes that begin library prep, as well as a unique barcode that tags all the reads that come from that cell.
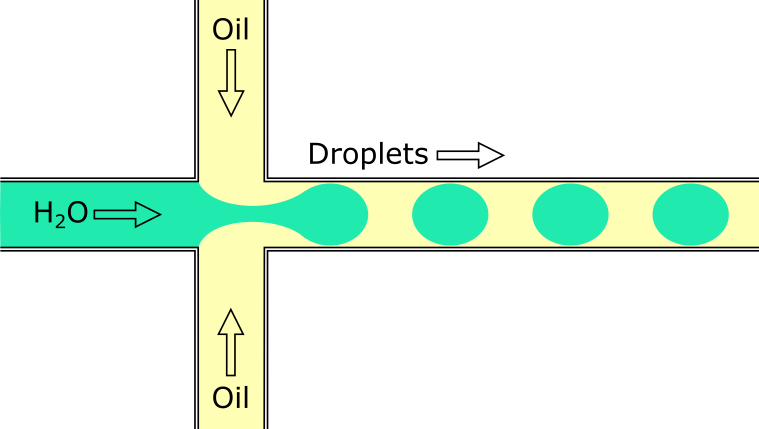
Image from https://en.wikipedia.org/wiki/Droplet-based_microfluidics#/media/File:Flow_Focusing_Device.png.
- Pool the cells and sequence.
Because each bead contained unique barcodes, we can pool all the cells back together and trace back the read to the cell it originated from. ✅
Let’s move on to sequencing protocols.
Sequencing protocols
There are two main sequencing protocols that are used in scRNA-seq.
- Full-length
- Tag-based
Full length sequencing protocols aim to sequence the whole transcript, while tag-based protocols capture either the 3’ or 5’ end.
Full length sequencing protocols in scRNA-seq are the same as in bulk RNA-seq.
In tag-based protocols, a unique molecular identifier (UMI) is attached to one end of the sequencing read.
This UMI is a 12 nucleotide long random sequence of bases.
The resulting transcript should now have-
- The nucleotide sequence.
- A unique barcode indicating the cell it originated from.
- A unique molecular identifier (UMI) identifying that specific molecule.
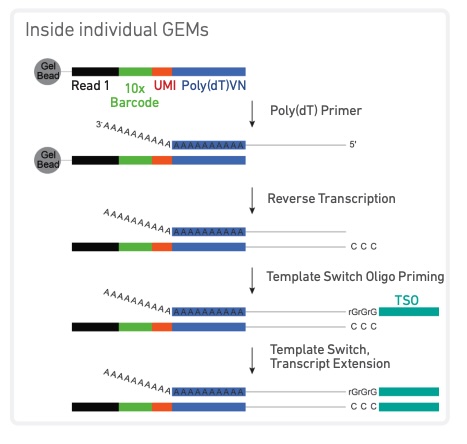
Image from https://assets.ctfassets.net/an68im79xiti/1C16trEdzy1Folq5xbOijE/7e6fb1f504e130bd561d898384da99d9/CG000315_ChromiumNextGEMSingleCell3-_GeneExpression_v3.1_DualIndex__RevB.pdf.
There are a few advantages to using the tag-based sequencing protocol.
More cost effective.
Because we are only sequencing the 3’ end (although can be either), we reduce the amount of sequencing required per cell.
Greater accuracy
The UMIs allow us to tag and identify individual transcripts. Allowing us to more accuratly quantify gene expression by distinguishing between original RNA transcripts and PCR duplicates.
Higher throughput
The UMI’s allow us to process millions of cells in a single sequencing run which increases the throughput, effeciency, and scalability.
But, we can’t have our cake and eat it too. It does come with a few disadvantages.
Less information about the transcript
Because we are only sequencing one end of the transcript (and not the entire transcript), it becomes more difficult to identify isoforms.
Less confidence in aligning reads.
Along those same lines, because we aren’t sequencing the entire transcript our ability to confidently align reads back to the genome is reduced.
Outro
And that’s it! In the next article we are going to dive more deeply into the data and considerations you have to take when processing it.
Then we are going to go into more downstream analysis.
If you want an in-depth explanation and a walkthrough of a common analysis pipeline, this course from the Sanger Institute is a great resource.
I hope you found this useful and see you in the next one!
